1. Elucidate the molecular mechanisms of repeat RNA metabolisms
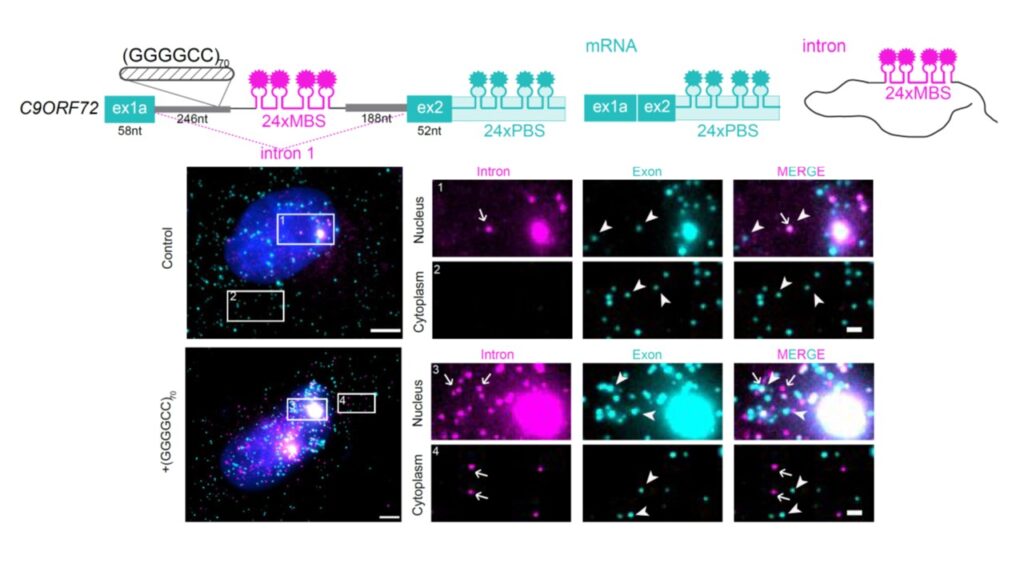
Nucleotide repeat expansions underlie a diverse group of neurological and neuromuscular disorders. The hexanucleotide GGGGCC repeat expansion located in the first intron of the C9ORF72 gene is the most common cause of both ALS and FTD. The leading hypothesis for the disease mechanism is gain of toxicity from the expanded repeats, which are transcribed in both sense and antisense directions, forming distinct sets of intranuclear RNA foci that may disrupt functions of RNA-binding proteins. An alternative but not mutually exclusive mechanism is the aberrant accumulation of dipeptide repeat (DPR) proteins produced by repeat-associated non-ATG (RAN) translation in all the six reading frames of both strands. Understanding the metabolism of repeat RNAs is crucial to addressing the root cause of disease mechanisms.
We identified cap-independent RAN translation of C9ORF72 repeats from the spliced intron, which can be upregulated by various stress stimuli through eIF2α phosphorylation signaling pathway. This supports a feedforward loop with initial repeat-mediated toxicity enhancing RAN translation and subsequent production of additional poly-dipeptides through integrated stress response, thereby promoting progressive disease (Cheng et al. Nature Communications 9: 51). We used single molecule imaging approach to discover that the GGGGCC repeats stabilized the spliced intron in a circular form, and mediate the circular intron export and translation in cytoplasm (Wang et al. Nature Communications 12: 4908). We measured the translation dynamics of different DPR reading frames of the GGGGCC repeats, and demonstrated different translation elongation rates in different frames as well as frame shifting between frames that generate chimeric dipeptides (Latallo et al. Nature Communications 14:5581).
2. Unravel the dysregulation of global RNA metabolism and the mechanisms of RNA toxicity underlying neurodegeneration
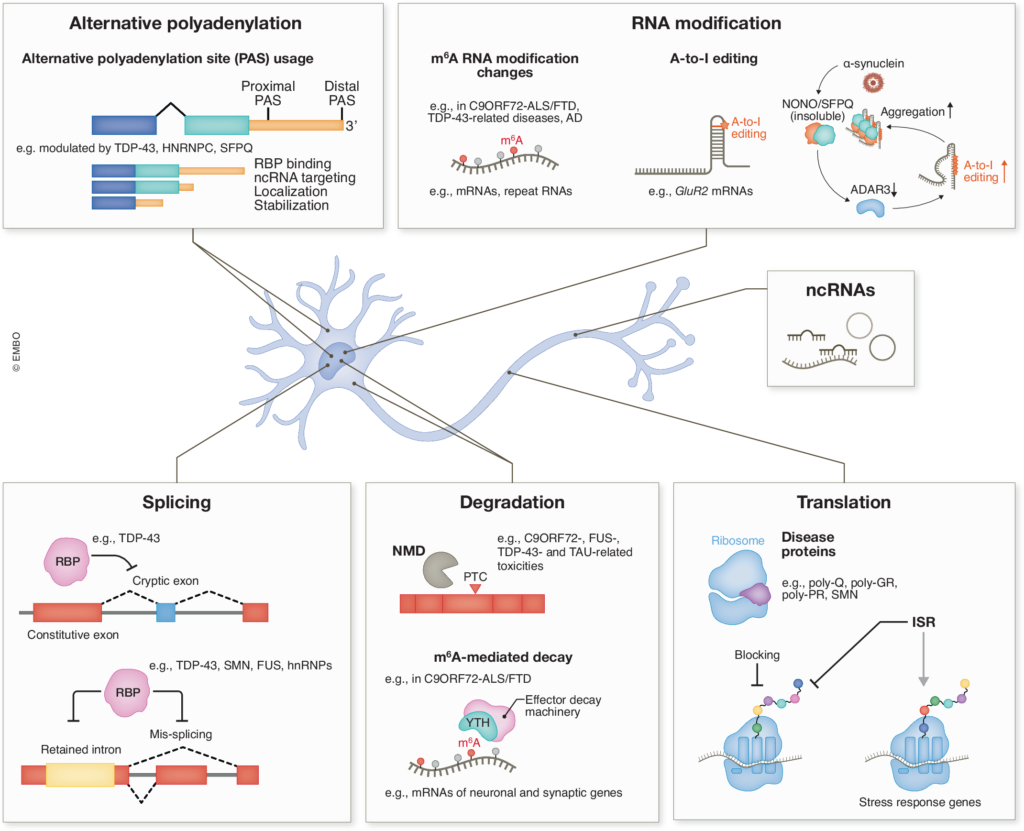
We are interested in uncovering the fundamental mechanisms and functions of RNA-binding proteins and RNA processing pathways in neurons, and how their dysfunction contributes to neurodegeneration. Our research encompasses RNA splicing, stability, modification, translation, and the roles of non-coding RNAs in epigenetics, etc. We believe these findings could shed light on the gene networks essential for maintaining proper neuronal function and health, and how dysregulation of RNA metabolism contributes to the selective vulnerability of neurons.
We found that N6-methyladenosine (m6A), the most prevalent internal mRNA modification, is downregulated in C9ORF72–ALS/FTD patient-derived neurons. This influences both global gene expression and repeat RNA metabolism. By elevating m6A methylation, we could significantly reduce repeat-derived toxic products from both sense and antisense strands, rescue global mRNA homeostasis, and improve patient neuron survival (Li et al. Nature Neuroscience, 26(8):8 132). We discovered that the (GGGGCC)n repeat RNA co-localizes with nuclear speckles and alters their phase separation properties. Together with aggregation of a nuclear speckle protein induced by poly-GR, this leads to impaired nuclear speckle integrity and global splicing defects in C9ORF72-ALS/FTD (Wu et al. Neuron, 112:3434). Additionally, poly-GR inhibits global translation by perturbing translation elongation, which increases ribosome collisions and ribotoxic stress response (RSR), promoting cell death (Dong et al. Science Signaling, 17:adl1030).
3. Identify disease modifiers by functional genomic screening
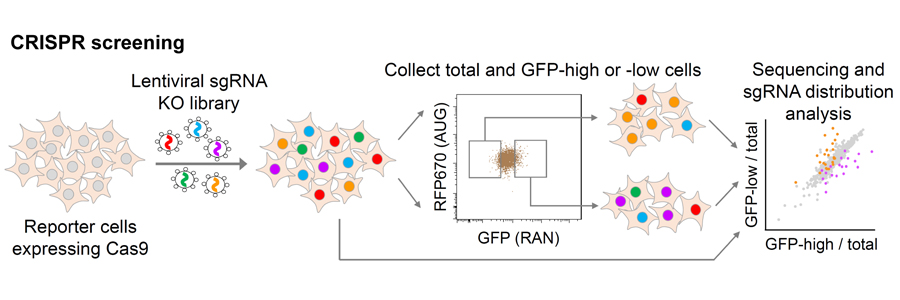
Genetic screenings in model organisms such as yeast, flies and nematodes have been instrumental in uncovering gene functions and understanding the pathogenic mechanisms induced by disease-causative mutations, thereby advancing the development of novel therapeutic targets. We now apply the cutting-edge CRISPR-Cas9 screening technology to identify modifiers directly in human cells and iPSC-derived neurons. We develop assays to screen for modifiers of specific molecular pathways (RNA processing, protein homeostasis), as well as factors that inhibit mutant gene-induced toxicity and enhance neuron survival. The identification of druggable gene products will pave the way for developing treatments for neurodegenerative diseases.
We used CRISPR-Cas9 knockout screens to identify genetic modifiers of poly-dipeptide production in human cells. We identified the RNA helicase DDX3X as a suppressor of RAN translation and elevating DDX3X expression can decrease DPR levels, rescue nucleocytoplasmic transport abnormalities and improve survival of C9ORF72-ALS/FTD patient iPSC-differentiated neurons (Cheng et al. Neuron 104:1-14). We also identified m6A writer proteins as modifiers of the DPR level. the m6A modification in the C9ORF72 intron sequence upstream of the expanded repeats enhances RNA decay via the nuclear reader YTHDC1, and the antisense RNA repeats can also be regulated. By elevating m6A methylation, we can significantly reduce the accumulation of potentially toxic products from both RNA strands and alleviate the disease phenotypes (Li et al. Nature Neuroscience, 26(8):8 132).
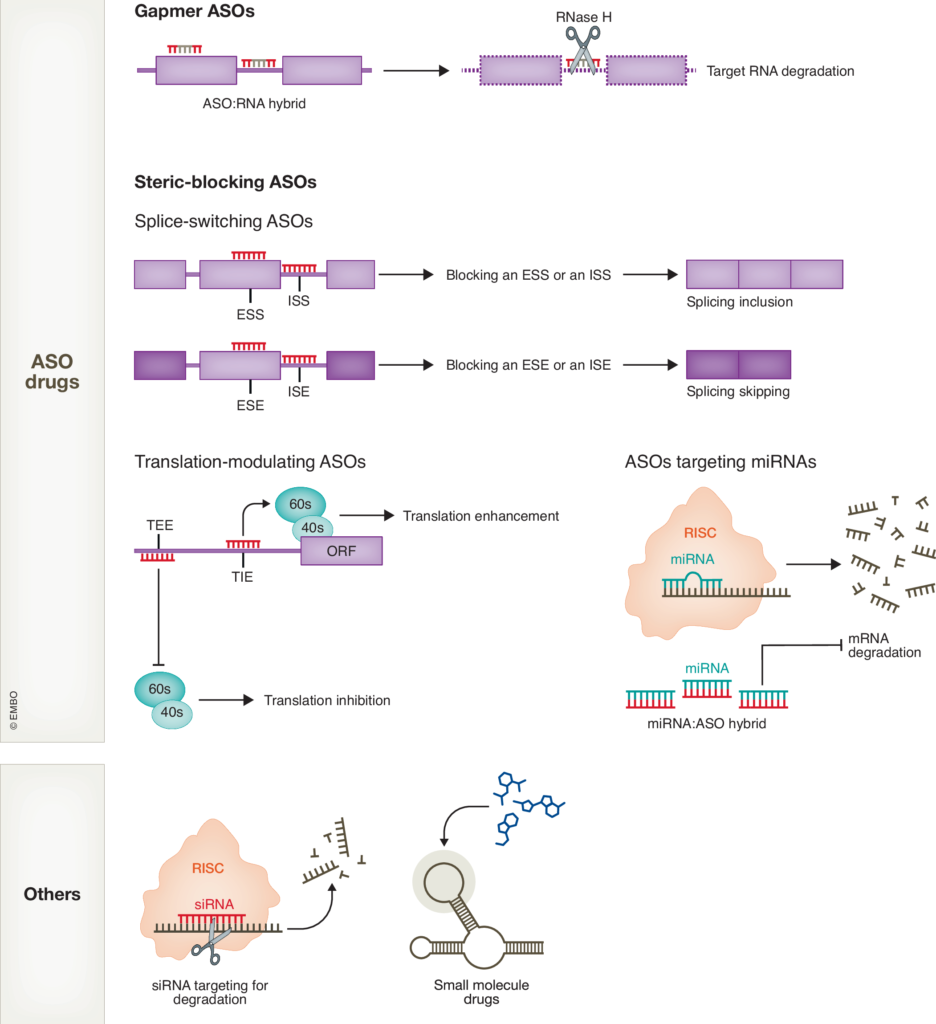
4. Develop RNA-targeting therapeutics
We aim to translate mechanistic findings into therapeutic development by leveraging antisense oligonucleotides (ASOs) and gene-editing approaches. ASOs are short, synthetic nucleic acids designed to bind specific RNA sequences, enabling targeted degradation, splicing modulation, or regulation of translation. These molecules offer a highly specific strategy to reduce the expression of toxic RNA or protein products, correct aberrant RNA processing events, or target modifiers of disease pathogenesis.
Additionally, we employ advanced gene-editing tools to manipulate RNA or DNA targets, including approaches such as base editing and prime editing to correct mutations, as well as RNA-targeting CRISPR variants to regulate RNA expression and function. These cutting-edge technologies hold great promise for developing precise and effective therapies for neurodegenerative diseases.